
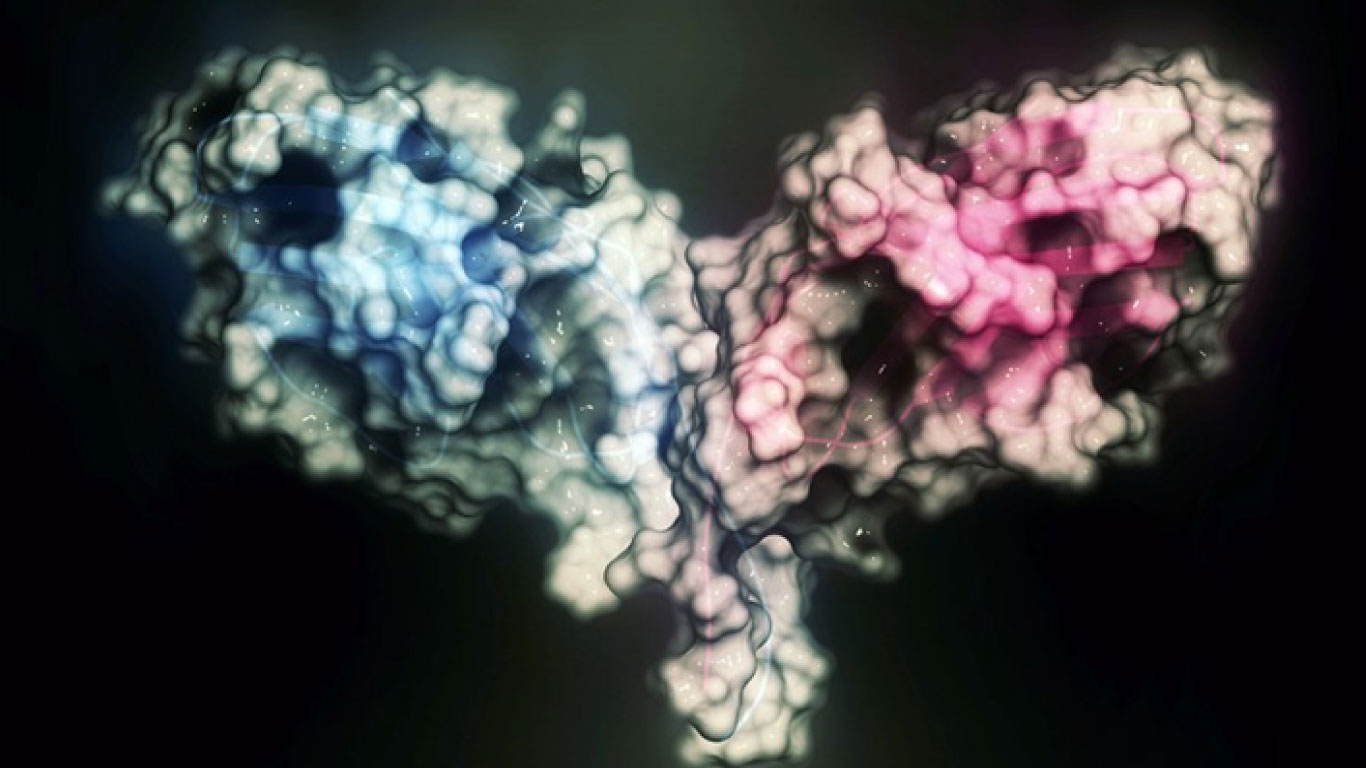
Un atlas de código abierto generado por una herramienta de IA llamada ESMFold2 aumenta enormemente el universo de proteínas conocido.
Publicado en: Nature noticias | 27 de mayo de 2026, Corrección del 27 de mayo de 2026
Por Ewen Callaway yMiryam Naddaf
El universo conocido de proteínas se ha ampliado considerablemente. Una herramienta de inteligencia artificial recientemente publicada ha generado un atlas con más de mil millones de estructuras proteicas predichas y miles de millones de secuencias proteicas adicionales.
La base de datos, conocida como ESM Atlas, fue presentada hoy por investigadores del Biohub de la Iniciativa Chan Zuckerberg, un instituto biomédico creado en San Francisco, California, por el fundador de Facebook, Mark Zuckerberg, y su esposa, la médica y educadora Priscilla Chan.
Este atlas supera a la base de datos AlphaFold de estructuras proteicas predichas en más de 800 millones de entradas, y a un atlas ESM anterior en unos 300 millones.
Las predicciones se realizaron utilizando ESMFold2, un modelo de IA que, según Biohub, supera el rendimiento de AlphaFold3, la última versión del sistema de Google DeepMind, y de otras IA de predicción de estructuras proteicas. El atlas se describe en un informe publicado hoy. “Este atlas muestra la totalidad de la biología de las proteínas, y especialmente las partes más desconocidas”, afirma Alex Rives, director científico de Biohub y líder del proyecto.
“Creemos que será una base muy valiosa para el descubrimiento de nuevos aspectos de la biología”.
Otros científicos están impresionados con los resultados, especialmente porque ESMFold2 es completamente de código abierto. Sin embargo, el modelo Biohub se adentra en un campo cada vez más competitivo, donde los modelos de proteínas, tanto de código abierto como propietarios, avanzan a pasos agigantados.
Predicciones de anticuerpos
ESMFold2 se basa en un modelo de “lenguaje de proteínas” que el equipo de Rives presentó en 2024, entrenado con miles de millones de proteínas de todo el árbol de la vida. Incluye secuencias “metagenómicas” del suelo, el océano y otros entornos, que no están presentes en la base de datos AlphaFold de estructuras proteicas predichas.
El equipo de Rives afirma que ESMFold2 supera a los métodos existentes, incluido AlphaFold3, a la hora de determinar la estructura correcta de los complejos de proteínas que interactúan, incluidas las moléculas de anticuerpos que se unen a sus dianas moleculares de antígenos.
En el informe, los investigadores describen cómo utilizaron ESMFold2 para diseñar nuevos anticuerpos y otras proteínas capaces de unirse firmemente a proteínas implicadas en cánceres y enfermedades inmunológicas. Al crearse y probarse en el laboratorio, una alta proporción de los diseños funcionó según lo previsto.
El equipo de Rives utilizó la herramienta para crear un atlas que contiene 1100 millones de estructuras proteicas predichas, así como información sobre las secuencias de 6800 millones de proteínas. La mayoría de estas provienen de secuencias metagenómicas que no han sido caracterizadas por completo. Rives espera que el atlas, que será de acceso libre, ayude a los científicos a establecer conexiones entre las partes conocidas y desconocidas del universo proteico. Utilizando el atlas, los investigadores encontraron similitudes estructurales entre las proteínas de defensa microbiana CRISPR y una proteína de edición genética identificada en un hongo del suelo en 2023 y encontrada en otras especies eucariotas².
Base de datos complementaria
El atlas recién publicado será «un recurso extraordinario para la biología», afirma Gemma Atkinson, bióloga computacional de la Universidad de Lund en Suecia. «Es fascinante ver cómo los modelos de lenguaje de proteínas a gran escala pueden capturar reglas fundamentales de la biología de las proteínas».
Christine Orengo, bióloga computacional del University College de Londres, afirma que las predicciones podrían ayudar a descubrir nuevos plegamientos y funciones de las proteínas, con implicaciones para el diseño de proteínas y la comprensión básica de la biología. Pero primero deberán ser evaluadas.
Martin Steinegger, biólogo computacional de la Universidad Nacional de Seúl, afirma que su mayor interrogante es qué tan bien ESMFold2 puede predecir la estructura de proteínas que difieren de las ya conocidas. Su equipo descubrió que la primera versión de ESMFold no era especialmente buena para predecir estructuras proteicas inusuales, especialmente aquellas encontradas en datos de metagenoma1.
El biólogo computacional Sergey Ovchinnikov, del Instituto Tecnológico de Massachusetts (MIT) en Cambridge, considera que el Atlas ESM es un complemento a la base de datos AlphaFold, ampliamente utilizada y que contiene más de 200 millones de estructuras de proteínas, en lugar de un sustituto.
Las predicciones de ESMFold2 sobre las proteínas que interactúan son impresionantes, añade Ovchinnikov, pero no del todo sorprendentes. A principios de este año, Isomorphic Labs, la filial biofarmacéutica de Google DeepMind con sede en Londres, presentó un modelo propio que logró avances sustanciales en la predicción de dichas estructuras. Los modelos de código abierto con los que el equipo de Biohub no comparó directamente ESMFold2 también han obtenido resultados impresionantes en la predicción de interacciones proteicas, afirma Ovchinnikov.
Según Ovchinnikov, la naturaleza totalmente de código abierto de ESMFold2, sin restricciones para su uso comercial, significa que podría tener una amplia acogida. «Creo que mucha gente estará encantada de probar ESMFold2».
doi: https://doi.org/10.1038/d41586-026-01686-3
Actualizaciones y correcciones
- Corrección del 27 de mayo de 2026: El Biohub de la Iniciativa Chan Zuckerberg se denomina simplemente Biohub, no CZI-Biohub.
Referencias
- Yeo, J. et al. Preprintin bioRxiv https://doi.org/10.1101/2025.04.23.650224 (2025).
- Saito, M. et al. Nature620, 660–668 (2023).Google Académico


