
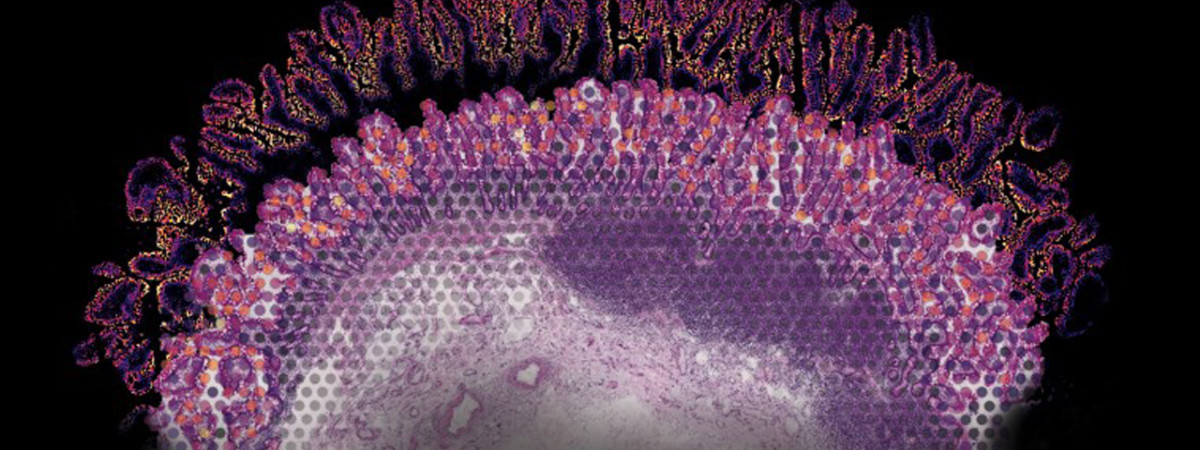
Los métodos computacionales y experimentales están acercando a los investigadores a su objetivo de revelar exactamente dónde se expresa cada gen en una célula o tejido.
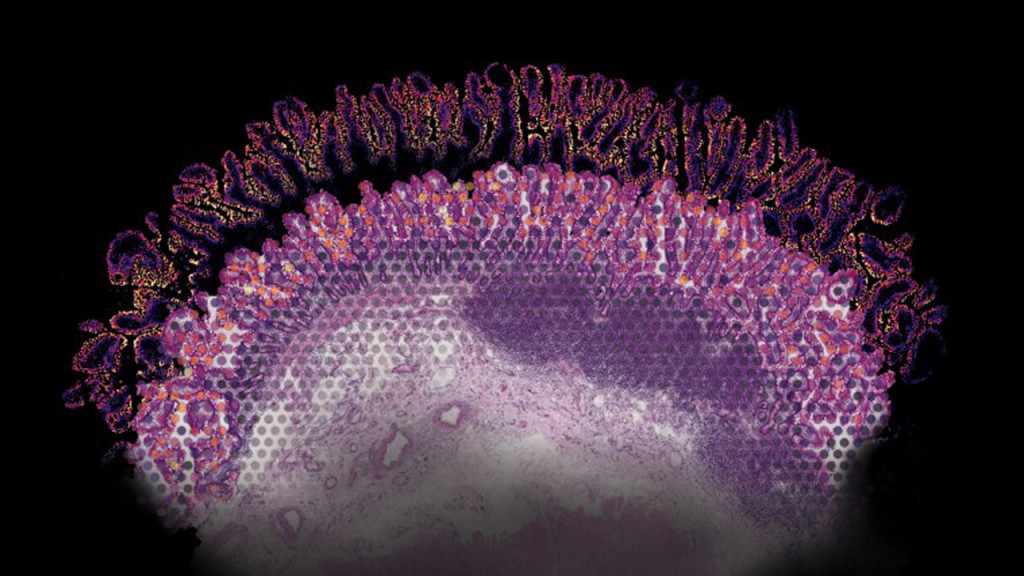
Al microscopio, los tejidos de los mamíferos revelan su intrincada y elegante arquitectura. Pero si se observa el mismo tejido después de la formación de un tumor, se verá un caos. Itai Yanai, biólogo computacional de la Facultad de Medicina Grossman de la Universidad de Nueva York, en la ciudad de Nueva York, está tratando de encontrar orden en este caos. “Hay una lógica particular en la forma en que se organizan las cosas, y la transcriptómica espacial nos está ayudando a verla”, afirma.
La “transcriptómica espacial” es un término general que abarca más de una docena de técnicas para trazar patrones de expresión génica a escala del genoma en muestras de tejido, desarrolladas para complementar las técnicas de secuenciación de ARN de células individuales. Sin embargo, estos métodos de secuenciación de células individuales tienen un inconveniente: pueden perfilar rápidamente el contenido de ARN mensajero (o transcriptoma) de grandes cantidades de células individuales, pero generalmente requieren la alteración física del tejido original, lo que sacrifica información crucial sobre cómo se organizan las células y puede alterarlas de maneras que podrían enturbiar análisis posteriores. El inmunólogo Ido Amit, del Instituto de Ciencias Weizmann en Rehovot, Israel, dice que tales experimentos a veces harían que su grupo cuestionara sus resultados. “¿Es este realmente el estado in situ, o simplemente estamos viendo algo que no es un factor importante o incluso no es real en absoluto?”.
En cambio, la transcriptómica espacial permite a los investigadores estudiar la expresión genética en muestras intactas, abriendo fronteras en la investigación del cáncer y revelando la biología hasta entonces inaccesible de tejidos bien caracterizados. Los «atlas» de información espacial resultantes pueden indicar a los científicos qué células componen cada tejido, cómo están organizadas y cómo se comunican. Pero compilar esos atlas no es fácil, porque los métodos de transcriptómica espacial generalmente representan una tensión entre dos objetivos en pugna: una cobertura más amplia del transcriptoma y una resolución espacial más estricta. Los avances en métodos experimentales y computacionales están ayudando ahora a los investigadores a equilibrar esos objetivos y, en el proceso, a mejorar la resolución celular.
Descamación de pescado
Los orígenes de la transcriptómica espacial se remontan a la década de 1960 y al desarrollo de la hibridación in situ . Esta técnica utiliza fragmentos marcados de ácido nucleico como sondas para detectar la presencia y la posición de secuencias complementarias de ADN o ARN en células o tejidos. Al principio, los investigadores utilizaban marcadores radiactivos, pero más tarde recurrieron a marcadores fluorescentes que pueden visualizarse con un microscopio.
En 1998, gracias a los avances en microscopía y procesamiento de imágenes, los investigadores pudieron identificar moléculas individuales de ARN en las células. Con este método de hibridación in situ con fluorescencia de una sola molécula (smFISH), fue posible visualizar transcripciones individuales de ARNm de varios genes simultáneamente utilizando sondas de diferentes colores. Pero las primeras versiones de smFISH podían monitorear solo tres o cuatro genes a la vez, muy por debajo de las decenas de miles de genes expresados en el transcriptoma humano. “Una de las limitaciones fundamentales de la microscopía es que no se pueden observar tantos colores o moléculas a la vez, aunque se obtenga esta información espacial realmente rica”, explica Fei Chen, biólogo celular del Instituto Broad del MIT y Harvard en Cambridge, Massachusetts.
Desde entonces, ingeniosas modificaciones de la técnica han superado esos límites. Por ejemplo, la técnica de hibridación in situ de fluorescencia multiplexada y robusta a los errores (MERFISH, por sus siglas en inglés), informada por la biofísica Xiaowei Zhuang y sus colegas de la Universidad de Harvard en 2015, permite detectar y discriminar entre miles de transcripciones de ARNm de diferentes genes utilizando solo unas pocas etiquetas fluorescentes1. A cada transcripción se le asigna un código de barras binario único compuesto de unos y ceros, y luego se etiqueta con múltiples “sondas de codificación” complementarias que contienen secuencias de lectura. Luego, las muestras pasan por rondas secuenciales de hibridación y obtención de imágenes con varias “sondas de lectura” marcadas con fluorescencia para descifrar este código de barras.
Cuando una sonda de lectura se une a la secuencia de lectura de una sonda codificadora y emite una señal fluorescente, se lee como un «1»; si no hay fluorescencia, se lee como un «0». Múltiples rondas de obtención de imágenes producen un código de barras binario que puede identificar el ARN detectado. La parte «robusto a errores» de la técnica se refiere al diseño de los códigos de barras: son lo suficientemente diferentes entre sí como para que haya pocas posibilidades de malinterpretar qué secuencia de ARNm se está detectando.
Aunque el método se describió inicialmente como una herramienta para el análisis de células individuales, el equipo de Zhuang también lo aplica a tejidos, incluido el cerebro humano2. “Al perfilar la expresión de 4.000 genes, pudimos generar un atlas celular definido molecularmente y resuelto espacialmente de la corteza humana con una resolución molecular y espacial sin precedentes”, sostiene. Este análisis, estableció la identidad y ubicación de más de 100 subtipos celulares distintos, y reveló diferencias sorprendentes en la composición celular y la organización de las estructuras cerebrales corticales en humanos en relación con los ratones. En trabajos anteriores, el grupo de Zhuang también ha utilizado la técnica para trazar partes del cerebro del ratón, incluida la corteza motora y el hipotálamo.
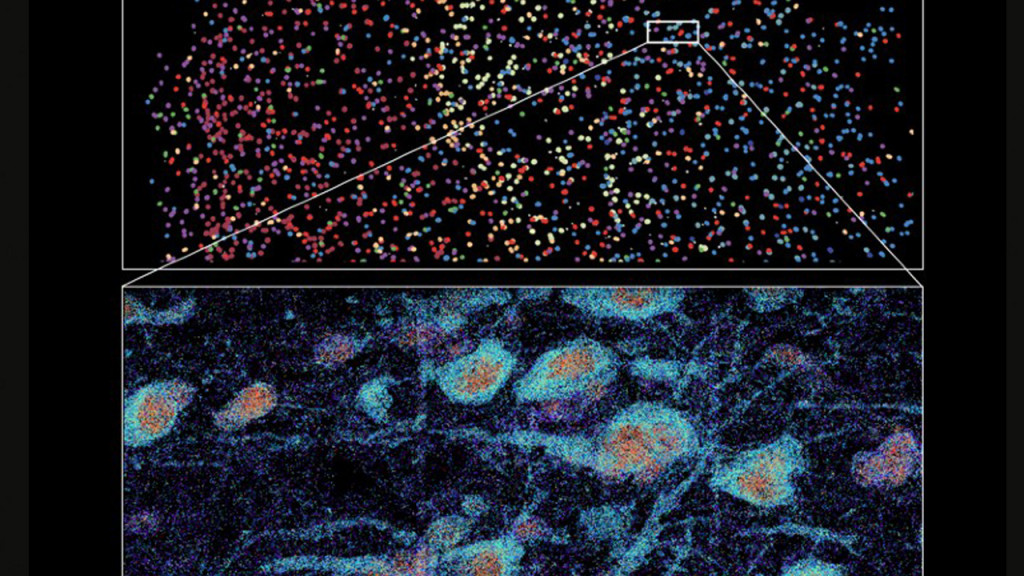
Otros métodos de codificación de barras y obtención de imágenes ofrecen beneficios similares. Por ejemplo, el mapeo de lectura de amplicones de transcripción con resolución espacial (STARmap), descrito por un equipo de la Universidad de Stanford en California en 2018, utiliza una forma de secuenciación in situ para detectar transcripciones de ARNm en muestras de tejido intactas3. Aprovechando un conjunto de códigos de barras específicos de genes, cada uno compuesto por 5 nucleótidos, el equipo de Stanford mapeó y cuantificó más de 1000 transcripciones de genes en tejido cerebral de ratón con resolución de una sola célula.
Pero los métodos basados en imágenes también tienen desventajas. Por ejemplo, a medida que estos enfoques se expanden para abarcar más objetivos, se vuelven cada vez más laboriosos. MERFISH puede detectar más de 10.000 genes a la vez, pero los experimentos a esta escala generalmente necesitan un paso adicional: un «protocolo de expansión de tejido» para aumentar el volumen de cada muestra de modo que la microscopía pueda resolver con éxito diferentes moléculas. Otro método, seqFISH+, supera esta limitación utilizando una estrategia de codificación por colores más compleja4. Pero seqFISH+ requiere muchas más rondas de etiquetado y obtención de imágenes (80, en comparación con las 23 de MERFISH) para el mismo número de genes. Y ambos métodos requieren más de un día de tiempo de microscopía ininterrumpido para recopilar datos a escala del transcriptoma.
Una gama de alternativas
Tal vez la limitación más fundamental de las técnicas basadas en hibridación es que los investigadores deben decidir de antemano qué genes desean atacar. “Una vez que se empiezan a seleccionar marcadores, se pierde información”, afirma Amit. Los métodos basados en matrices ofrecen una visión más amplia del transcriptoma, pero a un precio: tienen una sensibilidad menor y una resolución espacial reducida.
Joakim Lundeberg, genetista molecular del Instituto Real de Tecnología KTH en Estocolmo, uno de los pioneros de la transcriptómica espacial, describió este enfoque en 20165. Él y sus colegas salpicaron un portaobjetos de vidrio con una matriz ordenada de oligonucleótidos diseñados para capturar cadenas de ARNm. Estos funcionan uniéndose a la larga cola de nucleótidos de adenina que termina cada transcripción de ARNm. Después de aplicar una fina rebanada de tejido en la parte superior del portaobjetos, los investigadores trataron el tejido con sustancias químicas que lo hicieron permeable, lo que permitió que el ARN se filtrara y se uniera a la matriz. El ARN capturado luego se convirtió en ADN y se secuenció. Debido a que cada oligonucleótido contiene un código de barras distintivo que denota su posición en el portaobjetos, los datos finales revelan no solo la identidad del ARNm, sino también su ubicación en el tejido. Los datos resultantes pueden luego visualizarse como un mapa pixelado superpuesto en una imagen microscópica, en el que cada píxel revela qué genes se expresaron en cada posición.
El equipo de Lundeberg ha utilizado esta técnica para muestrear los transcriptomas completos de muestras de tejido cerebral y tumoral, aunque con una resolución espacial limitada. En el método original, los píxeles describían puntos de aproximadamente 100 micrómetros de diámetro, diez veces más anchos que una célula típica. Desde entonces, la técnica ha sido comercializada por la empresa 10x Genomics en Pleasanton, California, como la plataforma Visium Spatial Gene Expression, con un tamaño de punto de 55 µm. El equipo de Yanai ha utilizado la plataforma para mapear la arquitectura de tumores de páncreas y piel. E incluso sin resolución de una sola célula, han obtenido información valiosa sobre la arquitectura tumoral y las interacciones biológicamente importantes entre las células cancerosas, el tejido huésped sano y las poblaciones de células inmunes, dice.
En los últimos años se ha producido un gran esfuerzo para mejorar la resolución de los métodos basados en matrices. Por ejemplo, Chen y su colaborador Evan Macosko del Broad Institute desarrollaron un método llamado Slide-seq, que tiene una resolución de 10 µm (aproximadamente el tamaño de una célula), afirma Chen. La empresa 10x Genomics ha anunciado que su plataforma Visium HD de próxima generación, que se lanzará a finales de este año, también proporcionará una resolución de una sola célula, aunque hasta ahora no se han publicado datos.
En mayo, investigadores de la empresa de ciencias de la vida BGI-Shenzhen en Shenzhen, China, describieron un método basado en matrices que rompe la barrera de las células individuales7. Llamada Stereo-seq, utiliza matrices estampadas de nano bolas de ADN con código de barras que tienen aproximadamente 200 nanómetros de diámetro y unos pocos cientos de nanómetros de distancia. “En realidad tenemos algo así como 400 puntos de datos para generar una célula”, dice Xun Xu, director ejecutivo del Grupo BGI y uno de los desarrolladores del método. Se puede aplicar a muestras grandes, incluido un cerebro de macaco completo que se cortó en rodajas de tres por cinco centímetros, como se informó en una preimpresión ese año8. La secuenciación sola tomó casi dos meses, dice Ao Chen en BGI-Shenzhen, quien también es parte del equipo Stereo-seq.
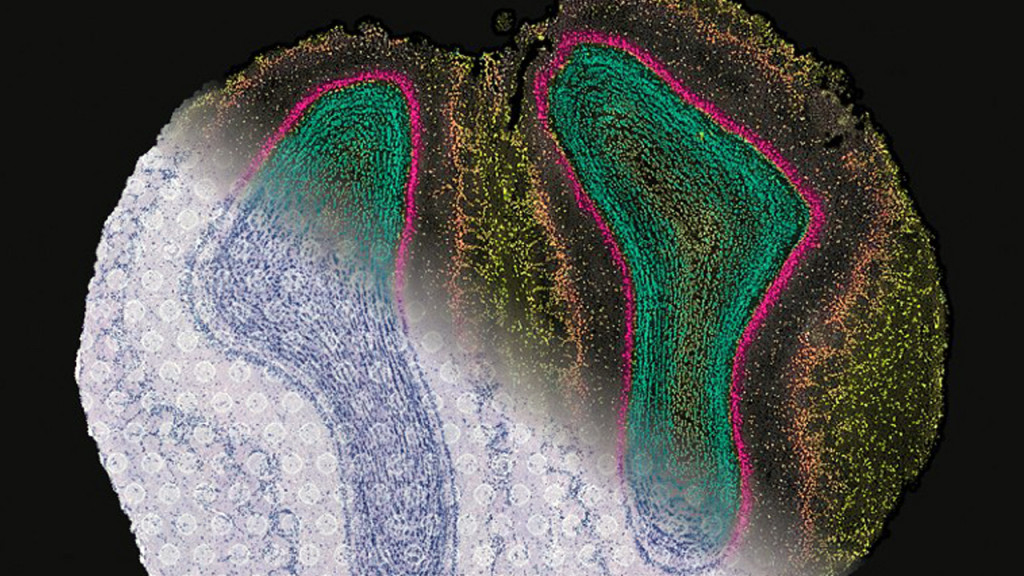
Pero a medida que la resolución se hace más estricta, también lo hacen los desafíos técnicos. Uno de ellos es la difusión: cuando los ARNm se filtran fuera del tejido, pueden propagarse lateralmente antes de encontrarse con una sonda de captura, lo que distorsiona los datos. Lundeberg dice que al optimizar el grado de permeabilización del tejido, los investigadores pueden limitar esta difusión a unos pocos micrómetros, lo que es más que suficiente para la resolución celular. “Si realmente desea ver la resolución subcelular, debería optar por las plataformas basadas en imágenes”, sugiere.
Otro desafío es de tipo físico: a medida que el tamaño de los píxeles disminuye, también lo hace el número de sondas disponibles para capturar el ARNm. Lundeberg dice que abandonó una versión de alta resolución de la plataforma de su grupo porque carecía de la sensibilidad necesaria para capturar señales de ARNm biológicamente relevantes. El equipo de BGI informa que Stereo-seq puede detectar normalmente entre 300 y 500 genes por célula, lo que ofrece una visión útil (aunque limitada) de la actividad de expresión génica. Aun así, el equipo ha utilizado el método para construir atlas en 3D que representan gráficamente los cambios espaciales en la expresión génica que acompañan al desarrollo embrionario en ratones7, moscas9 y peces cebra10.
Leyendo entre líneas
Para interpretar los datos espaciales se necesitan herramientas computacionales especializadas. Por ejemplo, los investigadores podrían necesitar deducir qué tipos de células están presentes utilizando datos que solo muestrean un subconjunto del transcriptoma. Muchos investigadores logran esto mediante el análisis paralelo de datos de secuenciación de ARN de células individuales recolectados del mismo tejido. “Entonces, se puede hacer coincidir y alinear lo que se está viendo en los datos espaciales con lo que se está viendo en los datos de células individuales”, sostiene Fei Chen. Esta comparación permite a los investigadores ubicar los tipos de células inferidos a partir de conjuntos de datos de secuenciación de ARN en mapas transcriptómicos espaciales.
Algunos algoritmos pueden incluso calcular la composición celular de los píxeles relativamente grandes producidos por plataformas como Visium, que pueden contener múltiples células. Fei Chen y el biólogo computacional de Harvard Rafael Irizarry desarrollaron un algoritmo de código abierto llamado descomposición robusta de tipos de células (RCTD) para este proceso de separación, también conocido como desconvolución puntual11. La RCTD es ampliamente aplicable a la mayoría de los métodos basados en matrices, dice Fei Chen. No solo identifica qué células están presentes en un píxel determinado, sino que también revela detalles faltantes sobre la actividad de expresión génica de esas células. La RCTD se puede aplicar a métodos basados en imágenes como MERFISH para la segmentación, agrega Fei Chen, identificando límites celulares a partir de datos de expresión génica derivados de la secuenciación de ARN de una sola célula.
Los datos de imágenes también pueden ser un recurso poderoso para la desconvolución celular, y la mayoría de las técnicas de transcriptómica espacial basadas en matrices pueden capturar dichos datos en paralelo, dice Mingyao Li, genómica estadística de la Universidad de Pensilvania en Filadelfia. “Se puede hacer zoom, se pueden ver las características específicas del tejido, cuántas células hay, cuál es la densidad celular y cuáles son las características morfológicas de las células individuales”, dice. Pero unir estos elementos es una tarea desafiante y que requiere una gran cantidad de datos, que a menudo requiere enfoques computacionales sofisticados.
Por ejemplo, Lundeberg y sus colegas publicaron un estudio12 en el que entrenaron un algoritmo de aprendizaje profundo con datos transcriptómicos e histológicos de un instrumento de Visium para extrapolar detalles que van mucho más allá del contenido de los puntos individuales. “Pudimos predecir con mucha precisión la expresión genética entre puntos”, dice, refiriéndose a los espacios físicos que son inherentes a todo método basado en matrices. “De hecho, pudimos inferir la resolución de una sola célula a partir de eso”.
Sin embargo, la identificación de los tipos de células es sólo el principio. Los diferentes tipos de células pueden tener fenotipos sorprendentemente distintos según su ubicación en un tejido, y estos patrones de expresión genética diferencial pueden hacer que un atlas celular espacial sea mucho más potente. Los algoritmos de aprendizaje automático también son útiles para desentrañar esta variabilidad. Por ejemplo, Amit y sus colegas desarrollaron una herramienta llamada DestVI que resuelve qué células están ubicadas en cada punto de la matriz y captura estados biológicos distintivos en varios tipos de células 13 . Utilizándola, el equipo identificó fenotipos de células inmunes en tejidos cancerosos. “Se puede llegar a un nivel mucho más alto de comprensión de la fisiología o patología en un tejido”, dice Amit.
Uniéndolo todo
Aunque parezca sorprendente, en un campo que produce tantos datos, lo que los investigadores de la transcriptómica espacial necesitan ahora son más datos. Iniciativas como el Atlas de células humanas, que ha publicado datos transcriptómicos recopilados de millones de células de 33 órganos ( www.humancellatlas.org ), son particularmente valiosas. Estos datos estandarizados y de alta calidad podrían utilizarse para entrenar algoritmos analíticos, por ejemplo.
La transcriptómica espacial aún no ha alcanzado el nivel de colaboración y compartición de datos que se observa en campos más establecidos, como la genómica o la transcriptómica de células individuales, y esto puede ser una fuente de frustración. En muchos casos, dice Fei Chen, los laboratorios solo comparten el mínimo requerido por los editores y los financiadores (los datos sin procesar de un experimento), lo que significa que podría llevar meses reproducir el trabajo. Pero ha habido avances prometedores. Tras la publicación de su trabajo Stereo-seq, por ejemplo, el Grupo BGI lanzó el Consorcio de Ómica Espacial Temporal, que ya ha atraído a más de 80 investigadores de todo el mundo. Su objetivo es utilizar varios métodos espaciales para abordar preguntas difíciles en áreas relacionadas con la fisiología humana, la patogénesis y la biología evolutiva.
Mientras tanto, los investigadores están tratando de mejorar aún más la tecnología. Por ejemplo, el equipo de Lundeberg está utilizando la transcriptómica espacial para inferir los cambios genómicos que ocurren durante el desarrollo del tumor de próstata, información que normalmente solo sería accesible a partir de la secuenciación del genoma de células aisladas. “En una sola sección de tejido, se ven estos eventos extremadamente tempranos que nadie ha buscado”, dice, y agrega que muchos de estos cambios se están produciendo en células que de otro modo parecerían benignas.
En cuanto a Yanai, está entusiasmado con la oportunidad de espiar cómo las células adyacentes se comunican y se influyen entre sí. Esta comunicación cruzada es un componente esencial de la formación y el desarrollo normal de los órganos, y podría ayudar a revelar los principios organizativos del tejido tumoral. “Las células cancerosas están manipulando a las células no cancerosas”, dice Yanai. La transcriptómica espacial podría capturar esa manipulación mientras ocurre. “Es como una pieza faltante del rompecabezas”, sostiene.
Nature 606 , 1036-1038 (2022)
Documento: https://doi.org/10.1038/d41586-022-01743-7
Referencias
1. Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. Science 348, aaa6090 (2015). Article PubMed Google Scholar
2. Fang, R. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.11.01.466826 (2021).
3. Wang, X. et al. Science 361, aat5691 (2018). Article Google Scholar
4. Eng, C.-H. L. et al. Nature 568, 235–239 (2019). Article PubMed Google Scholar
5. Ståhl, P. L. et al. Science 353, 78–82 (2016). Article PubMed Google Scholar
6. Stickels, R. R. et al. Nature Biotechnol. 39, 313–319 (2021). Article PubMed Google Scholar
7. Chen, A. et al. Cell 185, 1777–1792 (2022). Article PubMed Google Scholar
8. Chen, A. et al. Preprint at bioRxiv https://doi.org/10.1101/2022.03.23.485448 (2022).
9. Wang, M. et al. Dev. Cell 57, 1271–1283 (2022). Article PubMed Google Scholar
10. Liu, C. et al. Dev. Cell 57, 1284-1298 (2022). Article PubMed Google Scholar
11. C’able, D. M. et al. Nature Biotechnol. 40, 517–526 (2022). Article PubMed Google Scholar
12. Bergenstråhle, L. et al. Nature Biotechnol. 40, 476–479 (2022). Article PubMed Google Scholar
13. Lopez, R. et al. Nature Biotechnol. https://doi.org/10.1038/s41587-022-01272-8 (2022). Article Google Scholar
Download references ()


